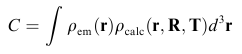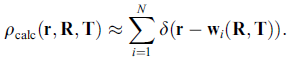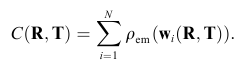| Tutorial - Manual Docking and Correlation-Based Refinement |
|
Sculptor provides techniques to refine the docking position of an already fitted structure. Typically the initial docking position comes from visually positioning the high-resolution structure, using Sculptor or another 3D visualization program. The refinement tools in Sculptor allow the structure to drift into the next local maximum of a cross-correlation scoring function. The initial part of this tutorial is identical to the feature-based docking tutorial - it asks you to download and visualize the files in exactly the same way. If you also went through the first tutorial, please go directly to the "Manual Docking" section.
Data SetsIn a first step the data sets have to be loaded into Sculptor - please download the following files (right-click, "Save Link As"):
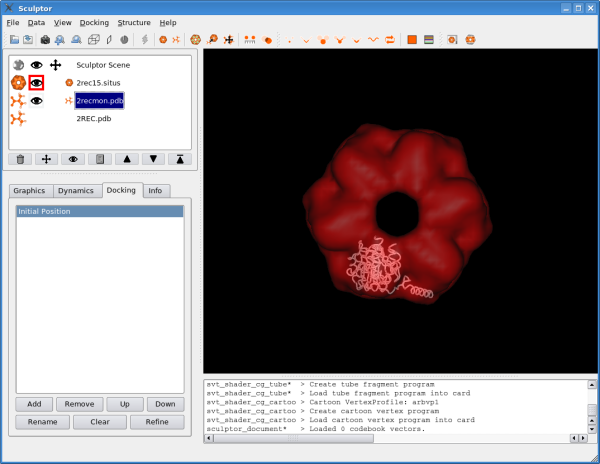
The atomic data can then be convolved with a Gaussian kernel to bring the resolution to a similar level as found in the volumetric data. This step is optional as the vector quantization procedure adjusts the level of detail anyway and as the correct resolution of a volumetric map is not trivial to determine. After downloading the data, please open them using the "file->open" menu-item. Hint: One can select multiple files in the file-open dialog and thereby load all three files at the same time. You should see now the three documents being listed in the top-left part of the main graphical user interface. Please adjust the order of the documents so that 2rec15.situs is the first document in the list - use the up and down arrows to reorder the entries if necessary. Your Sculptor window should now look like this:
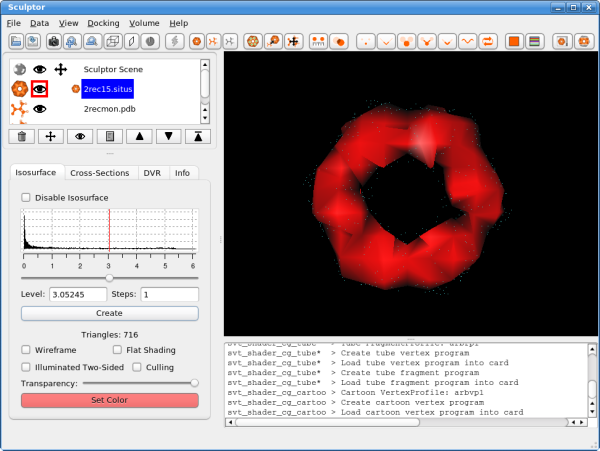
VisualizationThe visualization performed here is rather basic and does not show all the capabilities of Sculptor - please refer to the other documentation to get a more complete overview of the visualization features of Sculptor. Please start by clicking on the 2rec15.situs document. Now the main volume-rendering dialog appears right below the document list.
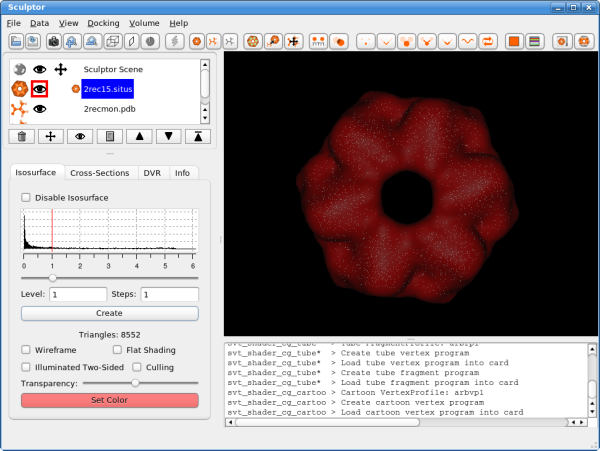
Click into the histogram to change the iso-surface threshold value. Adjust it to 1.0 (alternatively the level can also be typed in numerically - enter or clicking on the "Create" button will update the rendering). As the atomic structure needs to be docked into the volume, we have to render the low-resolution data in a transparent or semi-transparent way. This can be accomplished by either using the "Transparency" slider - moving it to the left makes the volume data more transparent. Another possibility is the "wireframe" mode.
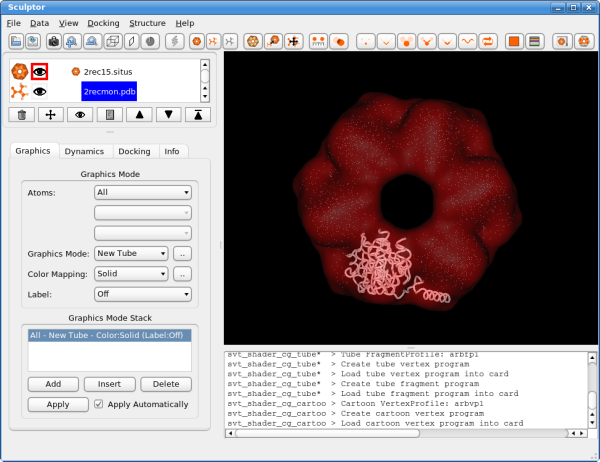
Now click on the next document in the list, the atomic structure of the monomer: 2recmon.pdb. The part just below the document list changes and shows now the rendering options for the atomic structures. Please select the "Graphics Mode" pull-down menu and select "New Tube". Please adjust also the color of the rendering by clicking on "Color Mapping" and setting it to "Solid". Now click on the button with the dots right next to "Color Mapping: Solid" field. A new color selector dialog is shown now and please select a bright color for the probe structure. You window should now look like the following:
Please click now also on the last document in the list, the structure of the entire hexamer "2REC.pdb", and adjust the rendering to "New Tube" and select a different color. As the "2REC.pdb" is only used for validation, we can now make it invisible by clicking on the eye symbol in the document list. The eye icon and the rendering of the hexameric structure should disappear.
Manual DockingSculptor allows you to manually dock a structure in a variety of ways. The most elegant solution requires a haptic device, and a later tutorial tells you how to manually dock a structure, if you have access to such a device. The tutorial here will explain how to use a standard 2D computer mouse. Select "2recmon.pdb" in the document list. Now click on the "Docking" tab. You should see now an empty list of transformations / docking positions. On the bottom there is a group of buttons - "Add", "Remove", "Up", "Down", etc. These buttons will help you to organize the different docking positions you find, being it the final docking positions of a oligomeric structure or just temporary solutions you just want to safe and compare. Please click on the "Add" button to add the initial start position to the list of transformations. Highlight the new entry and click on "Rename". Type in "Initial Position". Your Sculptor window should now look like this:
Moving objects in SculptorIn Sculptor the user can move the entire scene around, without
changing the positions and orientations of the objects relative to each
other. This is the normal mode of operation in Sculptor and is
reflected in the user interface by a little In this test case, the structure is already perfectly docked,
so now we will manually move it a bit around and try to dock it again
using the local refinement tools in Sculptor. Please click on the
little crosshair button, or alternatively one can also click right of
the eye icon in the "2recmon.pdb" line of the document list - in the
same column where the After misaligning the structure, please go back to the "Docking"-Tab and add another solution to the list by clicking on the "Add" button. Please rename the new solution to "New Position". By double-clicking on one the two solutions one can now quickly change the current position of the probe-molecule - useful to compare potential solutions with each other. You can try that using the newly created entries "Initial Position" and "New Position", but please make sure to double-click on "New Position" in the end to restore the slightly misaligned solution.
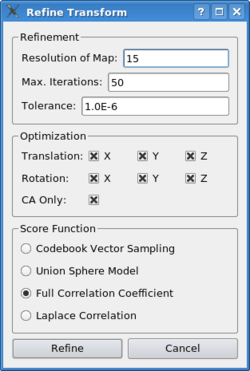
Refining Docking SolutionsPlease select/single-click the "New Position" entry in the list and click on "refine". The following dialog appears:
Sculptor offers a variety of established scoring functions that are optimized in a gradient-descent procedure to find the next local maximum. Most scoring functions need an estimation of the resolution of the target map - if in doubt, a slightly lower value will lead to a more robust behaviour, but in longer run-times. In the case of our example scenario, please input 15. The maximum number of iterations limits the run-time of the gradient-descent operation: Especially in cases where the current location of the probe still significantly deviates from the true solution, the algorithm might follow a gradient into the wrong direction and it might take a long time until it reaches a maximum score. Tolarance is the minimal difference between the score of two solutions at which the program will determine they are equal - the lower the value is, the longer it will take for the algorithm to converge. In the optimization box one can limit the degrees of freedom the procedure will manipulate - by default all translational and rotational degrees are enabled. If the probe molecule tends to drift away and one is not able to stabilize it in a certain region, a pure rotational refinement might be an option. With "CA Only" one can limit the refinement to the carbon-alpha atoms - useful in cases of very large structures where a full search over the entire molecule would be computationally too expensive.
Scoring FunctionsCurrently four different scoring functions can be selected. The most established criterion is the Correlation Coefficient:
The term rho_em represents the volumetric density of the cryo-em reconstruction and rho_calc the volume of the projected and blurred probe-structure. The same criterion is used in our full-automatic exhaustive search tool Colores. As rho_calc has to be recomputed for every change in the parameters R and T, the optimization based on the full correlation coefficient can be time-consuming for larger structures. Therefore we have developed two alternative scoring functions which are more efficient, the codebook-vector sampling and the union-sphere model. Codebook Vector Sampling is a very efficient reduced form of the correlation coefficient, based on feature-points which have to be pre-calculated before the refinement (please use the menu item "Docking->Feature Extraction->Neural Gas"). With the help of those feature vectors the term rho_calc can be reformulated into:
which then leads to a less complex version of the correlation function:
The Union Sphere Model is also an approximation of the correlation coefficient. Just like the full correlation it uses volume-data to model the probe structure, but to make it more efficient one assumes that the atoms have a uniform density. As a rule-of-thumb, the speed increases from Correlation Coefficient->Union Sphere->Codebook Vector Sampling, but the stability especially for intermediate to low-resolution maps might decrease (the faster algorithms might not find the correct docking location anymore and converge to an obviously wrong solution). If in doubt or if studying a smaller system, just choose the full correlation coefficient. The Laplacian Correlation is a full form of the correlation coefficient, but introduces the notion of a contour-based fitting. It utilizes a Laplacian edge-detection filter to distinguish between the contour and the interior of a volume data set. Only if contour and contour voxels and interior and interior voxels overlap in a fit, the score is maximized. This scoring criterion was introduced in Colores and has proven to be very successful to extended the viable resolution range for docking tools. This scoring function is recommended especially for intermediate- to low-resolution maps.
|